Recently, the research team led by Associate Professor Hailong Fan of our college made important progress in force-responsive polymer materials. Inspired by the cooperative regulation of mechanical responses by local sequence organization and dynamic noncovalent interactions in natural proteins, the team investigated a hydrophobic polyelectrolyte with statistically enriched adjacent cationic–aromatic motifs. Concentrated aqueous solutions of this polymer transform from viscous liquids into transient gel-like states under shear and recover fluidity after the removal of stress. This reversible shear-induced gel-like transition persists even after the polymer forms a dense associated structure in saline media. This work was published in Macromolecules. Yin An Chen of the College of Chemistry and Environmental Engineering, Shenzhen University, is the first author; Associate Professor Hailong Fan is the corresponding author; and the College of Chemistry and Environmental Engineering, Shenzhen University is the lead institution for the work.

Cation–π interactions play important roles in protein folding, molecular recognition, phase separation, and wet adhesion, and are often enabled by the local arrangement of cationic and aromatic groups. In aqueous environments, however, aromatic groups tend to aggregate hydrophobically, while hydration weakens or masks short-range cation–π interactions. Moreover, conventional copolymerization generally controls overall monomer composition rather than the local sequence distribution along the polymer chain.
To address this challenge, the team prepared P(ATAC-stat-PEA) through ideal copolymerization of the cationic monomer ATAC and the aromatic monomer PEA, whose reactivity ratios are close to unity. This strategy statistically enriches adjacent cationic–aromatic motifs along the polymer chain. The adjacent cationic units act as effective spacers that suppress direct hydrophobic aggregation of the aromatic groups in water while preserving the local geometry required for cation–π interactions.
Rheological measurements showed that the viscosity of concentrated aqueous P(ATAC-stat-PEA) solutions increased markedly with time under continued shear. The initially free-flowing solution gradually developed a transient gel-like state with a solid-like mechanical response. After shear was removed, the physically associated network relaxed and the sample recovered its fluidity. Repeated rheological cycling further confirmed that this transition is reversible and repeatable. In contrast, a nonaromatic control polymer, in which the aromatic monomer was replaced by a nonaromatic analogue, exhibited only ordinary shear thinning. These results indicate that the aromatic side group and the local cation–arene motif are key structural factors underlying this behavior.

Figure 1. Shear-induced reversible transient gel-like transition of P(ATAC-stat-PEA) in water.
The study further showed that the shear-induced gel-like transition depends strongly on both polymer concentration and shear rate. As the concentration increases, the characteristic interchain distance decreases and the degree of chain overlap/entanglement increases, making the system more susceptible to transient percolated network formation under shear. Small-angle X-ray scattering (SAXS) revealed concentration-dependent interchain correlations, while two-dimensional NOESY NMR spectroscopy showed short-range cation–arene proximity in the quiescent aqueous solution., providing a structural basis for shear-activated interchain association.
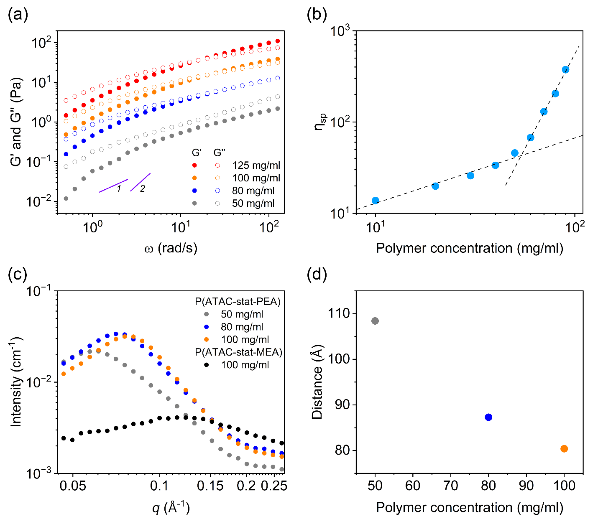

Figure 2. Concentration-dependent entangled state and shear-induced gelation kinetics of P(ATAC-stat-PEA) in water.
Upon salt addition, electrostatic screening drove the polymer into a denser associated structure, yet the reversible shear-induced gel-like transition persisted and showed a pronounced dependence on cation identity. Under the same experimental conditions, the gelation kinetics followed the order LiCl > NaCl > KCl. SAXS analysis also showed that the radii of the rodlike aggregates progressively decreased from LiCl to NaCl and KCl.
This trend is consistent with the known increase in aqueous cation–π affinity from Li+ to K+. Li+ is more strongly hydrated and less effective at competing for aromatic sites, thereby preserving more polymer-mediated interchain bridging. By contrast, K+ competes more effectively for aromatic sites and perturbs local polymeric cation–arene association more strongly. These results identify cation–π interactions as the dominant sequence-specific driving force for shear-induced gelation, with hydrophobic interactions contributing cooperatively.

Figure 3. Salt-dependent shear-induced gel-like transition and ion-regulated assembly of P(ATAC-stat-PEA) in saline media.
This study demonstrates that force-responsive behavior in synthetic polymers can be encoded not only by introducing specific functional groups, but also by controlling the local statistical organization of functional units along the polymer chain. Compared with complex precision sequence synthesis, conventional free-radical copolymerization provides a practical route to polymers with protein-like local motifs, offering a new design strategy for soft materials with reversible force responsiveness, adaptive rheology, and dynamic structural reconfiguration.
Article link:
https://doi.org/10.1021/acs.macromol.6c00991

 College of Chemistry and Environmental Engineering
College of Chemistry and Environmental Engineering